curlコマンドを利用すればネットワーク越しにファイルを取得できる
コマンドの使用形式は以下の通りである.
$ curl -O URL # リクエスト先のファイル名で出力. $ curl -o filename URL # 指定したファイル名で出力.
GENCODEから
ヒトとマウスの各種リファレンスデータを取得することができる.
NGSデータ解析の事前準備として以下のリファレンスデータを取得しておくとよい.

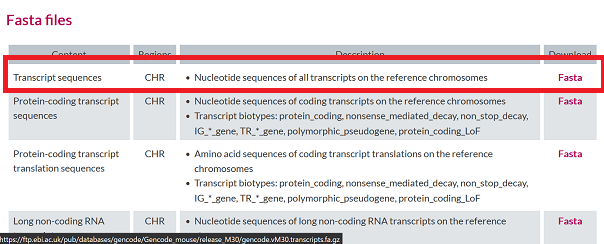
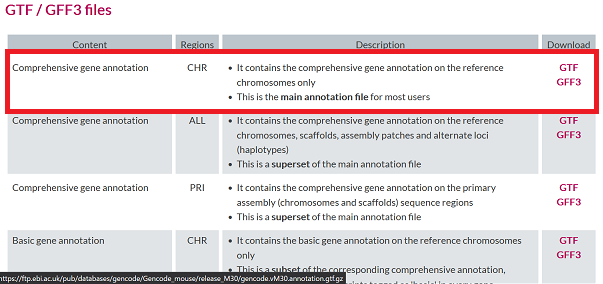
MUS=30 # Mouse Version: 30 HS=41 # Human Version: 41 URL_M=http://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_mouse URL_H=http://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human # GTF curl -O $URL_M/release_M$MUS/gencode.vM$MUS.annotation.gtf.gz #curl -O $URL_H/release_$HS/gencode.v$HS.annotation.gtf.gz # TRANSCRIPTOME #curl -O $URL_M/release_M$MUS/gencode.vM$MUS.transcripts.fa.gz #curl -O $URL_H/release_$HS/gencode.v$HS.transcripts.fa.gz # GENOME #curl -O $URL_M/release_M$MUS/GRCm39.primary_assembly.genome.fa.gz #curl -O $URL_H/release_$HS/GRCh38.primary_assembly.genome.fa.gz
downloadを実行する.
$ sh download_from_gencode.sh
gzという拡張子(*.gz)がついてるファイルは通常gzipで圧縮処理されたファイルである.
ファイルサイズの確認
$ ls -l gencode.vM30.annotation.gtf.gz
-rw-r--r-- 1 tom tom 29248040 10月 9 21:35 gencode.vM30.annotation.gtf.gz
$ ls -lh gencode.vM30.annotation.gtf.gz
-rw-r--r-- 1 tom tom 28M 10月 9 21:35 gencode.vM30.annotation.gtf.gz
$ cat gencode.vM30.annotation.gtf.gz | head # 圧縮ファイルなのでよく分からない.
$ gunzip gencode.vM30.annotation.gtf.gz
$ ls -lh gencode.vM30.annotation.gtf
-rw-r--r-- 1 tom tom 868M 10月 9 21:35 gencode.vM30.annotation.gtf
$ cat gencode.vM30.annotation.gtf | head # ちゃんと読めることを確認.
$ gzip gencode.vM30.annotation.gtf
$ zcat gencode.vM30.annotation.gtf.gz | head # zcatが利用できない場合はgunzip -c

GENCODEのGTFファイルフォーマットについては以下のURLから詳細を参照できる.
https://www.gencodegenes.org/pages/data_format.html
以下ではフォーマットの詳細が不明である場合を想定してファイル内を探索してみよう.
まずはheadコマンドを用いてデータ構造の概要を把握しよう.
$ zcat gencode.vM30.annotation.gtf.gz | head
##description: evidence-based annotation of the mouse genome (GRCm39), version M30 (Ensembl 107) ##provider: GENCODE ##contact: gencode-help@ebi.ac.uk ##format: gtf ##date: 2022-05-12 chr1 HAVANA gene 3143476 3144545 . + . gene_id "ENSMUSG00000102693.2"; gene_type "TEC"; gene_name "4933401J01Rik"; level 2; mgi_id "MGI:1918292"; havana_gene "OTTMUSG00000049935.1"; chr1 HAVANA transcript 3143476 3144545 . + . gene_id "ENSMUSG00000102693.2"; transcript_id "ENSMUST00000193812.2"; gene_type "TEC"; gene_name "4933401J01Rik"; transcript_type "TEC"; transcript_name "4933401J01Rik-201"; level 2; transcript_support_level "NA"; mgi_id "MGI:1918292"; tag "basic"; tag "Ensembl_canonical"; havana_gene "OTTMUSG00000049935.1"; havana_transcript "OTTMUST00000127109.1"; chr1 HAVANA exon 3143476 3144545 . + . gene_id "ENSMUSG00000102693.2"; transcript_id "ENSMUST00000193812.2"; gene_type "TEC"; gene_name "4933401J01Rik"; transcript_type "TEC"; transcript_name "4933401J01Rik-201"; exon_number 1; exon_id "ENSMUSE00001343744.2"; level 2; transcript_support_level "NA"; mgi_id "MGI:1918292"; tag "basic"; tag "Ensembl_canonical"; havana_gene "OTTMUSG00000049935.1"; havana_transcript "OTTMUST00000127109.1"; chr1 ENSEMBL gene 3172239 3172348 . + . gene_id "ENSMUSG00000064842.3"; gene_type "snRNA"; gene_name "Gm26206"; level 3; mgi_id "MGI:5455983"; chr1 ENSEMBL transcript 3172239 3172348 . + . gene_id "ENSMUSG00000064842.3"; transcript_id "ENSMUST00000082908.3"; gene_type "snRNA"; gene_name "Gm26206"; transcript_type "snRNA"; transcript_name "Gm26206-201"; level 3; transcript_support_level "NA"; mgi_id "MGI:5455983"; tag "basic"; tag "Ensembl_canonical";
##からはじまるコメント行の後に各エントリーが行ごとに配置されたタブ区切りの矩形データであることが分かる.
そこでまずコメント行を除いた後、1行目だけを取り出し、各列を行ごとに変換することで、各列のデータ種類を把握する.
$ zcat gencode.vM30.annotation.gtf.gz | grep -v '^#' | head -n 1 |
tr '\t' '\n' | awk '{print NR, $0}'
# grep -v '^#' で文頭にあるコメント行を除いている.
# ''又は""で囲むことで#のコメントとしての意味を失わせることができる.
# tr '\t' '\n'はタブ(\t)を改行(\n)に変換している.
# awk '{print NR, $0}' で行頭に行番号を附している.
1 chr1 2 HAVANA 3 gene 4 3143476 5 3144545 6 . 7 + 8 . 9 gene_id "ENSMUSG00000102693.2"; gene_type "TEC"; gene_name "4933401J01Rik"; level 2; mgi_id "MGI:1918292"; havana_gene "OTTMUSG00000049935.1";
3列目がgeneのデータを取得すれば全遺伝子の情報が取得できる.
抽出後の行数を算出し、
WEB上で公開されているSTATSデータと照合してみよう.
$ zcat gencode.vM30.annotation.gtf.gz | grep -v '^#' | awk -F'\t' '$3=="gene"' |
wc -l
# awk -F '\t' によりデフォルトのスペース・タブ区切りでなくタブ区切りのみであることを指定
56691
全遺伝子情報が取得できていることが確認出来たら、次にENSEMBL GENE IDと遺伝子シンボル名との対応表を作成してみよう.
$ zcat gencode.vM30.annotation.gtf.gz | grep -v '^#' | awk -F'\t' '$3=="gene"' |
awk -F '\t' '{print $9}' | awk -F ';' '{print $1, $3}' | awk '{print $2, $4}' |
tr -d '"' | sort | head
# GTFファイルの9列目に目的のENSEMBL IDとGene Symbleのデータがある.
ENSMUSG00000000001.5 Gnai3 ENSMUSG00000000003.16 Pbsn ENSMUSG00000000028.16 Cdc45 ENSMUSG00000000031.18 H19 ENSMUSG00000000037.18 Scml2 ENSMUSG00000000049.12 Apoh ENSMUSG00000000056.8 Narf ENSMUSG00000000058.7 Cav2 ENSMUSG00000000078.8 Klf6 ENSMUSG00000000085.17 Scmh1
FAQ| Table Browser| Table Browser User's Guide| FTP site explanation|
Fetch NCBI RefSeq gene information from Table Browser
| clade: | mammal |
| genome: | human |
| assembly: | GRCh38/hg38 |
| group: | Genes and gene predictions |
| track: | NCBI RefSeq |
| table: | RefSeq All(ncbiRefSeq) |